
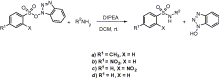


Acknowledgments
We are thankful to DST for XRD facility; CIF, IITG for NMR, ESI-MS facility; DST SERC for research grant (Sanction No. SR/FT/CS-011/2008, FAST TRACK SCHEME). PNB thanks IITG for fellowship.
The N-hydroxybenzotriazole (HOBt) sulfonate esters undergo amidation under ambient conditions in the presence of di-isopropylethyl amine. This method can be applied to varieties of amines including sterically hindered amino acid esters in less time with reasonably good yields.
HOBt ester of sulfonic acids can be a replacement for pentafluorophenyl and trichlorophenyl ester as well as chloride moiety of sulfonyl chloride for amidation.
Sulfonamide unit has drawn attention of synthetic organic chemists because of its diversified applications in medicinal chemistry1 as anticancer, antiinflammatory, antimicrobial, anticonvulsant, and antiviral agents.2 Another important property of sulfonamides is its crystalline nature, which renders easier handling, purification, and characterization. Furthermore, sulfonamides are very important catalysts in asymmetric synthesis.3 Moreover, tosylated derivatives are the reactive intermediates to synthesize various molecules including heterocycles containing N, O atoms.4 Therefore, finding out novel routes for their synthesis is important.
Although, there are many protocols for the synthesis of sulfonamides from sulfonyl chlorides,5 these methods are associated with many drawbacks, as sulfonyl chlorides are very reactive and difficult to handle. Therefore, it is important to develop a method in which sulfonic acids are activated by a better leaving group so that amidation is facilitated under ambient conditions. In this connection, Stephen group has contributed remarkably in replacing sulfonyl chlorides by activating sulfonic acids as corresponding pentaflurophenyl sulfonates6 and trichlorophenyl sulfonates.7 However, green chemistry practice restricts not to use polyhalogenated compounds, since they are perceived to be toxic and expensive. Moreover, said methods are associated with bases such as DBU, LHMDS, and NMP along with heating condition. In another report, benzotriazole (Bt) activation is described in which long reaction time and heating conditions are necessary.8
Our group is interested in converting amino acid esters into sulfonamides under milder conditions. Therefore, we wanted to find out an alternative to pentaflurophenyl sulfonates and trichlorophenyl sulfonates for synthesizing sulfonamides at ambient conditions without using any strong bases in order to address the limitations of earlier reported methods while retaining the benefits of activated sulfonate esters. We chose N-hydroxybenzotriazole (HOBt) ester of p-toluene sulfonic acid which is also as stable as pentaflurophenyl sulfonates and trichlorophenyl sulfonates. HOBt is largely used in peptide synthesis 9 as it acts as racemization suppressant. 10 Moreover −OBt group acts as a better leaving group for aminolysis.
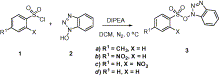
Preparation of HOBt ester of sulfonic acid (3) was achieved by the reaction of sulfonyl chloride and HOBt, in the presence of DIPEA (Scheme 1) following a reported protocol.11 Resulting white crystalline stable product in high yield inspired us for further investigation on aminolysis of this ester.

Synthesis of various sulfonate esters.
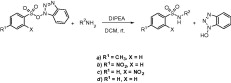
We noticed that when TsOBt was treated with benzyl amine at room temperature in the presence of 1 equiv of DIPEA, N-benzyl sulfonamide of p-toluene sulfonic acid (entry 1, Table 1) was produced in good yield ( Scheme 2), as white crystalline solid after usual acid base work-up and column chromatography. The present method is compatible with primary (entries 1–3 and 5–7), and secondary amines (entry 4) with comparable efficiency to that demonstrated in the previously reported methods. 6 and 7 All the reactions went to completion within 1–2.5 h except for entry 9.
Synthesis of sulfonamides from N-hydroxybenzotriazole esters of sulfonic acids
| Entry | Sulfonate ester | Amine | Timeb (h) | Isolated yielda (%) |
|---|---|---|---|---|
| 1 | R1 = CH3 X = H | Benzylamine | 1.5 | 75 |
| 2 | R1 = CH3 X = H | n-Butyl amine | 2.5 | 60 |
| 3 | R1 = CH3 X = H | α-Methyl benzyl amine | 2 | 84 |
| 4 | R1 = CH3 X = H | Piperidine | 2 | 91 |
| 5 | R1 = CH3 X = H | Cyclohexyl amine | 2.5 | 80 |
| 6 | R1 = H X = H | Cyclohexyl amine | 1 | 84 |
| 7 | R1 = NO2 X = H | Cyclohexyl amine | 1 | 90 |
| 8 | R1 = CH3 X = H | NH2-Phe-OMe | — | — |
| 9 | R1 = H X = H | NH2-Phe-OMe | 7 | 34 |
| 10 | R1 = NO2 X = H | NH2-Phe-OMec | 2.5 | 47 |
| 11 | R1 = CH3 X = H | NH2-Gly-OMe | 2 | 71 |
| 12 | R1 = NO2 X = H | NH2-Gly-OMe | 1.5 | 82 |
| 13 | R1 = H X = NO2 | NH2-Gly-OMe | 1.5 | 79 |
| 14 | R1 = CH3 X = H | NH2-Ala-OMe | 2.5 | 65 |
| 15 | R1 = NO2 X = H | NH2-Ala-OMe | 2.5 | 76 |
| 16 | R1 = H X = NO2 | NH2-Ala-OMe | 2.5 | 67 |
Yield after purification by passing through a short silica gel column.
TLC is checked for every 30 min.
Conversion was 60% only and few unidentified products were noticed.
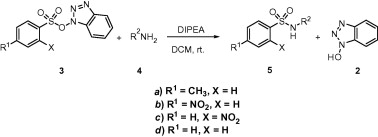
Synthesis of sulfonamides from TsOBt.
Amino acid esters also showed similar reactivity except for sterically hindered methyl ester of phenylalanine. Reaction of 3a with methyl ester of phenylalanine did not progress at all (entry 8). However, removal of the electron donating methyl group on benzene ring of the sulfonyl chloride showed better reactivity by yielding at least 34% although the reaction took longer time (7 h, entry 9). On the other hand, insertion of an electron withdrawing group increases the yield further (47%) and reduces the reaction time as well (2.5 h, entry 10). Difference in reactivity was also observed when cyclohexyl amine was used with variation of the substitution at para position of 3. When electron donating methyl group was present the reaction was slower (2.5 h, entry 5), the reaction time was reduced to 1 h when methyl group was removed (entry 6) and an electron withdrawing group was inserted (entry 7). Sequential increment of the reaction yield was also observed (entry 5–7). Such substituent effect also can be observed by comparing the percentage yield in entries 11 versus 12, and 14 versus 15, although the differences are not much but consistent. Variation of the position of the nitro group also influences the reaction yield which is clear from the comparison of the same in the entries 12 versus 13 and 15 versus 16. Therefore, in general, the scheme is influenced by the electronic factors on benzene ring of the sulfonate esters of N-hydroxy benzotriazole (3).
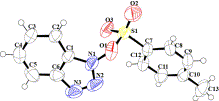
All the products that were new were characterized by 1H NMR, 13C NMR, IR, and ESI-MS and the reported products and activated esters were characterized by 1H NMR, IR, and ESI-MS. The data is matched with reported data. TsOBt (3a) is also characterized by single crystal XRD (Fig. 1, characterization data of all the reaction products including crystallographic data are provided in the Supplementary data in detail).
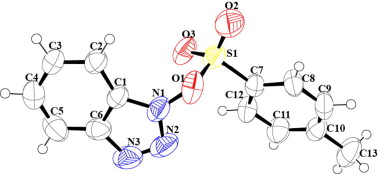
ORTEP diagram of TsOBt with 50% ellipsoid (CCDC # 834974).
In terms of reactivity, these activated HOBt esters undergo amidation at ambient conditions as it works for sulfonyl chlorides. This methodology eliminates the use of expensive solvents; rather DCM was used in most of the cases. However, sometimes sonication and addition of DMF were necessary for better solubility. This method offers many advantages over the existing methods for the synthesis of sulfonamides: (a) the harsh bases, for example, NMM, DBU were replaced with milder DIPEA, (b) long reaction times were reduced to 1–3 h, and (c) required reaction temperature was reduced to the ambient temperature. Moreover, the current method is free from the production of HCl, which enables it to be applicable to those substrates in which acid labile groups such as, Boc are present. Furthermore, as the base being used is milder, it can also be applied to those substrates in which base labile groups like Fmoc are present. In addition to the above advantages, N-hydroxy benzotriazole can be easily removed from the reaction mixture at the end by mere washing with water.
Thus, in this letter we have shown that N-hydroxy benzotriazole sulfonate motif is a potential replacement for the chloride unit of sulfonyl chloride for the synthesis of sulfonamides. Unlike the previous activation methods in which pentaflurophenyl and trichlorophenyl esters of sulfonic acids were used, current activation undergoes amidation quite easily without the use of solvents like NMP, high temperatures and harsh bases, retaining decent yields even in case of complicated amines, such as, hindered amino acid esters. Regarding the issue of cost, N-hydroxybenzotriazole is cheaper than pentafluorophenol, while comparable to trichlorophenol. Therefore, these properties enable N-hydroxy benzotriazole sulfonate to be a good synthetic auxiliary for sulfonamide synthesis.
We are thankful to DST for XRD facility; CIF, IITG for NMR, ESI-MS facility; DST SERC for research grant (Sanction No. SR/FT/CS-011/2008, FAST TRACK SCHEME). PNB thanks IITG for fellowship.

General procedures.
Science, 288 (2000), p. 2132
Comprehensive Medicinal Chemistry, Vol. 2Pergamon Press, Oxford (1990) Chapter 7.1
J. Med. Chem., 49 (2006), p. 4384
J. Am. Chem. Soc., 120 (1998), p. 10994
J. Med. Chem., 35 (1992), p. 2496
Med. Res. Rev., 5 (2003), p. 535
Curr. Med. Chem., 10 (2003), p. 925
Chem. Commun. (2010), p. 4589
Chem. Eur. J., 16 (2010), p. 8259
J. Org. Chem., 54 (1989), p. 906
J. Org. Chem., 51 (1986), p. 2386
Sulfonamide synthesis from tosyl chlorides using catalysts: Adv. Synth. Catal., 349 (2007), p. 1873
Synlett (2007), p. 2501
Tetrahedron Lett., 50 (2009), p. 1117
Using base: Green. Chem., 8 (2006), p. 835
Directly from thiols: J. Org. Chem., 74 (2009), p. 9287
J. Am. Chem. Soc., 126 (2004), p. 1024
Org. Lett., 4 (2002), p. 2549
Chem. Commun. (2007), p. 1074
J. Org. Chem., 69 (2004), p. 1849
Copyright © 2011 Elsevier Ltd. All rights reserved.





{kind=link}
{kind=link}
{kind=link}
{kind=link}