Screen reader users, click here to load entire articleThis
page uses JavaScript to progressively load the article content as a
user scrolls. Screen reader users, click the load entire article button
to bypass dynamically loaded article content.
ScienceDirect is phasing out support for older versions of
Internet Explorer on Jan 12, 2016. For the best product experience, we
recommend you upgrade to a
newer version of IE
or use a different browser: Firefox or Chrome.
For additional information please see the ScienceDirect Blog page.
Close
An
unexpected involvement of ethyl-2-cyano-2-(hydroxyimino) acetate
cleaved product in the promotion of the synthesis of nitriles from
aldoximes: a mechanistic perception
While
attempting to synthesize nitriles from aldoximes using O-sulfonate
esters of oxyma [ethyl 2-cyano-2-(hydroxyimino)acetate], an unexpected
involvement of oxyma cleaved product in promoting the synthesis of
nitriles was observed. Such involvement of the oxyma cleaved product in
the reaction mechanism, together with the usual anticipated pathway
improved drastically the applicability of the method by reducing the
time needed for the reaction to be completed over that of the sulfonyl
chlorides. Other advantages of the present protocol are excellent yields
in ambient and milder conditions.
Nitriles are important synthons in organic syntheses as they are involved in many functional group transformations.1
There are two essential general methods for their preparation. Firstly,
the nucleophilic substitution of alkyl halides/aryl halides with a
stoichiometric amount of inorganic cyanide resulting in alkyl/aryl
nitriles, respectively, which is frequently accompanied by the
elimination of hydrogen halides. Especially, alkyl and aromatic nitriles
(benzonitriles) can be synthesized by Sandmeyer reaction and
ammoxidation. Although α,β-unsaturated nitriles can be synthesized by
Wittig reaction of the corresponding aldehydes with cyanoalkyl
phosphate, the method often produces a mixture of E- and Z-isomeric nitriles. 2
The second one involves the conversion of an aldehyde into oximes and
dehydration of the latter into nitrile. The dehydration of aldoximes,
which are easily prepared from the corresponding aldehydes, is a
candidate for clean nitrile synthesis. Various procedures have been
reported using reactive species like N-chlorosuccinimide/PPh3, 3 thionyl chloride-benzotriazole, 4N-(p-toluenesulfonyl) imidazole (TsIm), 5 Vilsmeier reagent, 6 Burgess reagent, 7 cyanuric chloride, 8S,S-dimethyl dithiocarbonate, 9 sulfonyl chlorides, 10 and triflyl imidazole 11. Apart from these, several transition metal catalysts such as [RuCl2(p-cymene)]2/molecular sieves, 12 rhenium(VII)oxo complexes, 13 InCl3, 14 TiCl3(OTf), 15 and Cu salts, 16
have been used to carry out the same transformation which are expensive
and often associated with long heating conditions. Recently, there was a
report that describes the same transformation using TsOBt/DBU. 17 Though, it works well it is noteworthy that N-hydroxybenzotriazole is explosive on heating. In the present context, even though, the sulfonyl chlorides 10
were used for this transformation, longer reaction times were
necessary. We wish to report herein a new method for the said
transformation where aryl sulfonate esters of oxyma are used. A
mechanistic investigation is also performed. The increased reactivity of
the oxyma-O-sulfonates can be attributed to the unexpected involvement
of the oxyma-cleaved product and thereby enabling the reaction to be
completed much faster than its previous analogues. This is the first
report of such involvement of the oxyma-cleaved product in the reaction
mechanism.
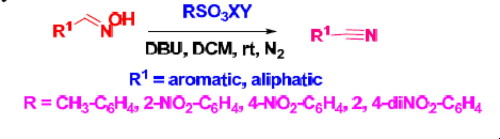
In continuation of our work on the promotion of activated sulfonate esters as reagents for various organic transformations,18
we want to enunciate the synthesis of nitriles from aldoximes using
sulfonate esters of oxyma which could be easily prepared from the
corresponding sulfonyl chloride and oxyma in the presence of DIPEA (Scheme 1).19
In order to verify the applicability of the sulfonate esters of oxyma
in nitrile synthesis, we have prepared 2,6-dichlorobenzaldoxime from
2,6-dichlorobenzaldehyde and hydroxyl amine hydrochloride and set our
objective to synthesize nitriles from respective aldoximes. We took
1 mmol of 2,6-dichlorobenzaldoxime and 1 mmol of sulfonate
ester of oxyma in the presence of 3 equiv of triethyl amine in
dichloromethane. Interestingly, the reaction was completed within
1 h and furnished the product in very good yield. Inspired by this
result, we went on to standardize the reaction.
Having
the proof of principle for the synthesis of nitriles from aldoximes
using the new reagents, we wanted to examine the potential of the
activated sulfonate esters. Out of all the sulfonate esters that we have
synthesized (IIIa–d), it was found that IIIa
have modest reactivity and took a little longer time. However the
insertion of the electron withdrawing group at the aromatic ring has
dramatically changed the reactivity and promoted the reaction within
10 min (IIIb–d).
The noteworthy point is that the reaction with all these activated
esters is so fast (10 min) that it was difficult to distinguish
their reactivity for this transformation in promoting the reaction and
yielding the product. However, we proceeded further to carry out this
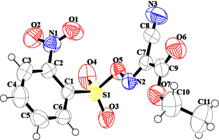
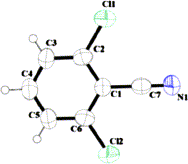
transformation using 2-NO2-C6H4-SO3XY (IIIc, Fig. 1).
Figure 1.
X-ray crystallographic structure of IIIc (ORTEP diagram with ellipsoid of 50% probability, CCDC No. 903881).
To determine the suitable base for this transformation, several bases were tried over the same substrate as shown in Scheme 2.
It was observed that in the presence of a tertiary amine base the
reaction went to completion smoothly and furnished the product in very
good yield in all the cases except with 2,4,6-collidine and
2,3,5-collidine (see Table S1 in Supplementary data).
DBU and DABCO took very less time. Therefore, we decided to proceed
further with DBU. It is important to note that the formation of the
product in the absence of any base was not observed within 10 min.
as it was found with DBU. Next, we envisaged the stoichiometry of the
base using different equivalents of DBU. We noted that 2.5 equiv
were compulsory for completion of the reaction. The conversion was not
complete with lesser amount of base and resulted in the desired product
in reduced yield (see Table S2 in Supplementary data).
However, the unreacted starting material could be recovered. From this,
it was clear that the ratio of the base to the substrate should be
1:2.5 so that the reaction gets completed within 10 min. Solvent
screening experiments revealed that CH2Cl2 was the best solvent for this transformation (see Table S3 in Supplementary data).
In methanol there was a side product corresponding to the competitive
reaction for the formation of the sulfonate ester of methanol.
The current methodology is applicable to a wide veriety of substrates (Table 1) and tolerates a wide variety of functional groups, for example NO2,
MeO–, EtO– etc. Under optimized conditions, all the substrates went to
completion within 10 min and yielded the product in very good
yield. There was no influence of electron donating groups, for example
MeO–, EtO–, Br, Cl etc. or electron withdrawing groups, for example NO2 on the yield and dynamics of the reaction.
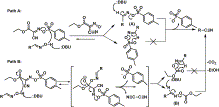
In
path A, base deprotonates the aldoxime to generate a nucleophile which
attacks the electrophilic centre over sulfur of the reagent IIIa and forms sulfonate ester of aldoxime as intermediate A. Finally, second equiv of the base deprotonates the carbon of the imine, thereby expels the –OSO2Ar
group which is a good leaving group and results in the generation of
the nitrile along with oxyma (by-product). We computed the charge
distribution by Mulliken analysis for compound IIIa at B3LYP/6-31G(D,P) level of theory21 and found that partial positive charge over the carbonyl carbon (0.59) of compound IIIa is much less than that over the sulfur center (1.29). Therefore, the path A is the logically anticipated route of the reaction.
In
path B, base deprotonates the aldoxime to generate a nucleophile, which
attacks the electrophilic centre over carbonyl carbon of the reagent IIIa to produce O-ethoxycarbonyl aldoxime as intermediate B
along with the corresponding sulfonic acid. The second equiv of the
base deprotonates the carbon of the imine and generates nitrile. Such
participation of the oxyma derived product in the reaction mechanism has
not been described before. The intermediates A and B were purified and characterized by 1H NMR, mass, and FT-IR (see Supplementary data for experimental details, characterization data and copies of the spectra). The intermediate B was further analyzed by single crystal XRD ( Fig. 3a).
However, the possibility of generation of the product by intramolecular
hydrogen abstraction from both of the intermediates (A and B) as shown in Scheme 3
cannot be ruled out, but that should be predominant in high dilution
and high temperature condition such as in gas phase reactions as it is
reported for intermediate A. 22
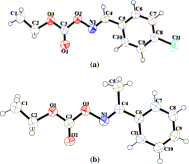
Figure 3.
X-ray crystallographic structure of (a) (E)-4-chlorobenzaldehyde-O-ethoxycarbonyloxime and (b) acetophenone-O-ethoxycarbonyloxime (ORTEP diagram with ellipsoid of 50% probability, CCDC No. 918220 and 918221, respectively).
In
order to precisely predict the ability of DBU whether it is capable of
deprotonating the ethylenic proton we have synthesized the authentic
intermediates A and B
with complete characterization and finally subjected to the reaction in
the presence and in the absence of 1 equiv of DBU. Interestingly,
it was found that the reaction completed in the presence of DBU in
10 min, whereas it did not progress in the absence of DBU (checked
until 12 h). Therefore, it can be concluded that the intramolecular
H-abstraction did not occur.
Furthermore,
to support the mechanism, the reaction was repeated using a ketoxime as
substrate and corresponding products to the intermediates A and B were
expected. However, O-ethoxycarbonylketoxime ( Fig. 3b)
was obtained as sole product with very good yield. This indicates that
in case of the ketoxime, either the reaction follows only path B or the
first step of path A is reversible. No oxyma could be recovered when
ketoxime was used, which proves the complete involvement of oxyma
cleaved product, whereas, approximately 50% of oxyma could be recovered
when aldoxime was used as the substrate which proves that some of the
cleaved product of the oxyma is involved in the promotion of the
reaction in yielding the desired nitrile quickly and with better yield
than its counterpart, that is sulfonyl chlorides. 10
In
conclusion, we have demonstrated a newer method for the synthesis of
nitriles from aldoximes under milder conditions using sulfonate esters
of oxyma. The unusual involvement of oxyma in promoting the reaction was
established with single crystal X-ray structure, which actually
promotes the reaction in much shorter times compared to that of the
sulfonyl chlorides. This is the first report of the involvement of an
oxyma derived product in the mechanism. This method has several
advantages over the existing methods as, (a) it precludes the usage of
the expensive reagents and toxic metals, (b) reaction times have been
shortened, (c) the reactions were carried out at lower temperatures
(room temperature), etc. Therefore, the current methodology finds large
applications in the synthesis of natural products where the tolerance of
a good number of other functional groups is in force.
Acknowledgments
We thank the CIF-IITG for NMR, LC–MS; DST (FIST) for single crystal XRD facility and the Department of Science & Technology, India
(FAST TRACK scheme, sanction no. SR/FT/CS-011/2008) for financial
support. D.D. and N.B.P. thank IIT-Guwahati for fellowship. We also
thank Mr. Harikrishna Sahu and Dr. Aditya Narayan Panda for
computational calculations. We also thank the reviewers for their
constructive suggestions.
Representative procedure for nitrile synthesis:
In an oven-dried two-necked 50 mL round-bottomed flask, equipped
with a stirring bar, a solution of the oxime (1.0 mmol) and 2-NO2-C6H4-SO3XY (1.5 mmol) dissolved in anhydrous CH2Cl2
(5.0 mL) was placed under the atmosphere of nitrogen. The reaction
mixture was stirred at room temperature for 5 min, then DBU
(2.5 mmol) was added drop wise over 2 min. The reaction
mixture became a clear homogeneous solution after addition of DBU. The
reaction was monitored by TLC. The reaction mixture was diluted with
EtOAc and washed with water (2 × 5 mL) followed by brine
(2 × 5 mL) upon complete consumption of the starting
material. Product was purified by column chromatography.
Neese,
F.; ORCA, version 2.9, An ab initio, DFT and semiempirical SCF-MO
package; Max-Planck Institute for Bioinorganic Chemistry, Mülheim a. d.
Ruhr, Germany, 2010.
The
complicated substrates that included the conjugated aldoxime (entries
11, 13, and 14) underwent the reaction to furnish the product in
reasonably good yields. The important advantage of this protocol is that
(entry 11) the stereochemistry is retained, that is E isomer of the aldehyde gave the E isomer of the corresponding nitrile unlike the Wittig reaction which gives a mixture of E and Z isomers. This reaction works well for aliphatic systems as well (entries 16–19). All the products have been characterized with 1H NMR, 13C NMR, IR, and GC–MS.20 Out of all these, the product of entry 2 was characterized with single crystal XRD also (Fig. 2).
Figure 2.
X-ray crystallographic structure of the product of entry 2 in Table 1 (ORTEP diagram with ellipsoid of 50% probability, CCDC No. 905250).
To investigate the mechanism of the reaction, we took 4-CH3–C6H4–SO3XY (IIIa)
as reagent and TEA as base to slow down the reaction. Premature
quenching (at 30 min) of the reaction, where 4-chlorobenzaldoxime
was used as substrate, resulted in two new spots in TLC, which were
finally characterized as the intermediates (E)-4-chlorobenzaldehyde-O-tosyloxime and (E)-4-chlorobenzaldehyde-O-ethoxycarbonyloxime (A and B in Scheme 3,
respectively). Existence of these two intermediates in the reaction
mixture indicates that the reaction may proceed following both of the
probable mechanisms as depicted in Scheme 3 as the path A and the path B.
Sulfonamide synthesis using N-hydroxybenzotriazole sulfonate: an alternative to pentafluorophenyl (PFP) and trichlorophenyl (TCP) esters of sulfonic acids

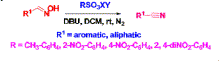
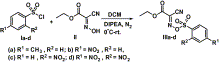
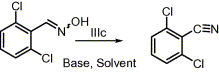






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}